David R. Powell,
Lloyd Allison &
Trevor I. Dix,
Computer Science and Software Engineering,
Monash University,
Australia 3800.[*]
We Define:
Global and local,
optimal and summed,
affine (linear) gap-costs (i.e. 3-state),modelling-alignment (M-alignment).
Compare it to:
Standard Smith-Waterman local-alignment with
significance-test based on shuffling and re-alignment.
Results:
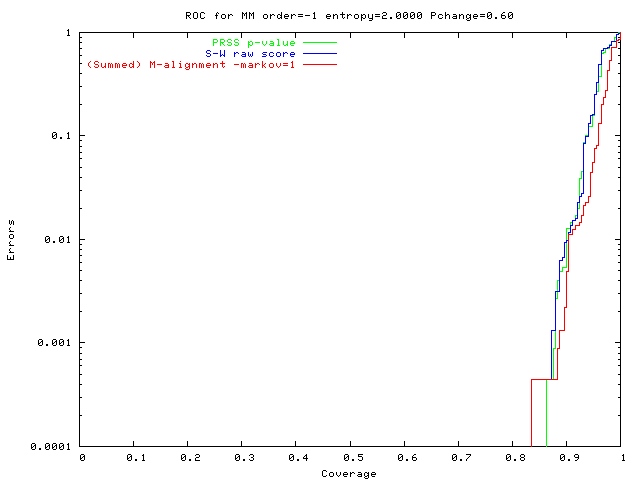
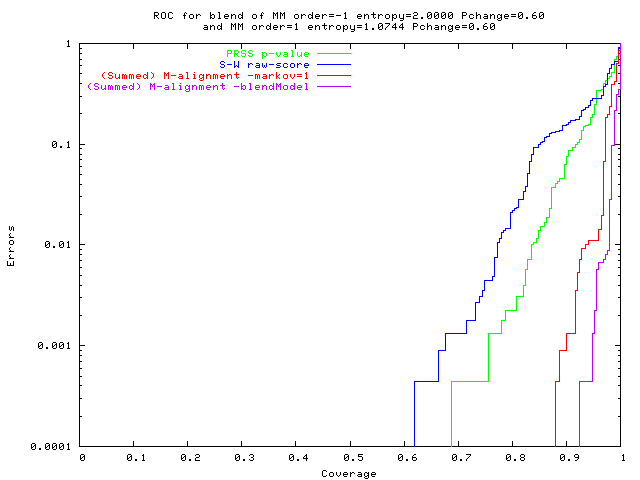
Both solve easy problems equally well.
M-alignment performs better on
compressible populations,
mixed populations, and
populations of mixed sequences.
A seminar to
1. Bioinformaticians who lunch,
MO23 Biology,
University of York, UK,
1.15, 25/11/2004,
2. eHealth Res. Centre (CSIRO +Qld. Gov),
300 Adelaide St., Brisbane, Qld., Australia,
9am, Fri. 27 May 2005,
3. School of Software Engineering and Data Communications,
QUT, Brisbane, Qld., Australia,
3pm, Fri. 27 May 2005.
Modelling-alignment (M-alignment) can (& should(!))
change (i) optimal alignments &
(ii) rank-order of matches against a data-base.
Can use it with almost any population model.
On easy problems (0-order data):
M-alignment ~ Smith Waterman + shuffling sig' test.
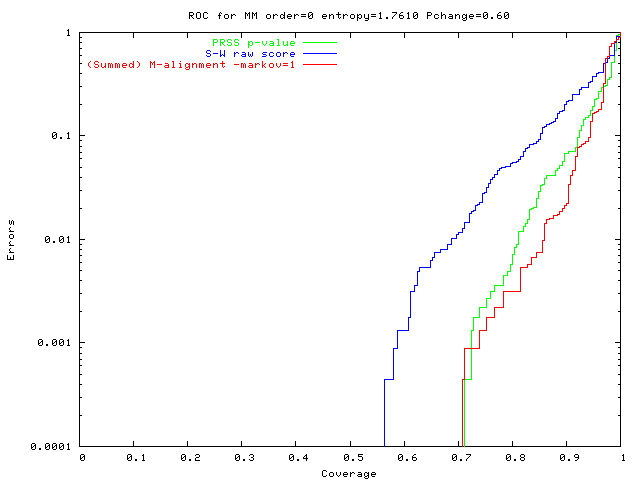
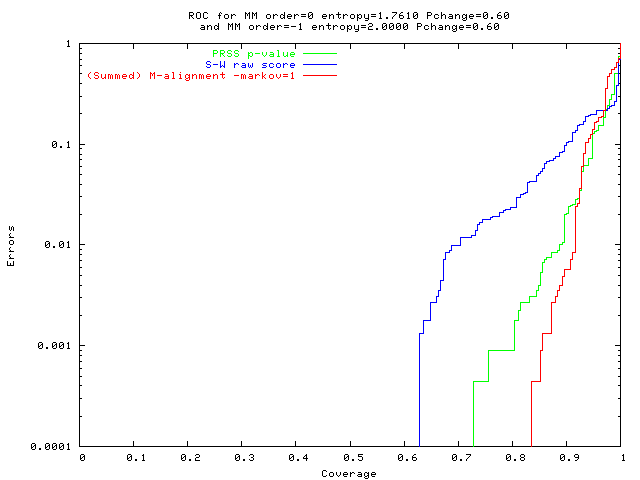
On hard problems:
M-alignment is much more accurate
(fewer false +ves,
fewer false -ves).
Reading:
L. Allison, D. Powell & T. I. Dix (1999).
Compression and approximate matching.The Computer Journal,42(1) pp1-10
-- Describes principle, compression.
Implements 1-state, global, optimal version.
D. R. Powell, L. Allison, & T. I. Dix (2004).
Modelling-Alignment for Non-Random Sequences.[*]
ACS Australian Joint Conf. on Artificial Intelligence (AI2004),
Cairns Queensland, pp.203-214, 6-10 Dec. 2004,
Springer-Verlag, LNCS/LNAI 3339, isbn:3540240594. [*]
Link also leads to the software.