-----JAVASCRIPT IS OFF-----
Compression and Analysis of Biological Sequences. Why and How.
On work with
Julie Bernal,
Minh Duc Cao,
Samira Jaeger,
Chris Mears,
David Powell,
Tim Edgoose,
Linda Stern* ,
Trevor Dix,
and others at
Monash U., .au., and
* Melbourne U..
Seminar presented to:
Computer Science, University of Wales, Aberystwyth,
Tuesday 16 Jan 2007,
Computing Sciences,
University of East Anglia (UEA), UK,
Wednesday 24 Jan 2007,
Mathematical Sciences Inst.,
Australian National University (ANU), Canberra,
Monday 26 Feb 2007,
and
Intelligent Systems Research Group,
FIT, Monash University,
Tuesday 13 March 2007,
Will discuss these related problems :
Compression of DNA.Compression of protein.Uses of information content in sequence analysis.Relating 2 or more (non-random) sequences.
-- Lloyd Allison users.monash.edu.au/~lloyd/Seminars/2007-BioCompression/index.shtml
Abstract
Compression can be rather addictive.
It has an obvious ``figure of merit'' --
the compressed size of some standard data-set, and
it is natural to strive to beat your competitors on this measure.
Biological sequences are hard to compress;
the more compression the better but there is usually limited value
in fighting over the third significant digit.
Other properties of a compression method can be just as important
or more important.
This talk gives some reasons why it is challenging, interesting and useful
to compress biological sequences.
It also presents two simple models for compressing biological sequences
(a possible sub-addiction in compression is to complicated models,
but simple is often good);
we get good results for DNA and protein.
Why Compress Biological Sequences?
Not to save time or space! (Not yet.)For analysis.Good model <=> good compression ( compression = modelling + coding ) Note, "masking-out" implies that
low-information content =
zero information.
Not true!
Desirable features of a model:
(Good compression.)Based on biological knowledge.Simple, few parameters.Can give per-symbol information content.Efficient algorithm.
DNA
Alphabet = {A, C, G, T}.Various kinds of repeat.Usually approximate.Reverse complementary repeats.
Protein
Amino acids.|Alphabet| = 20. (log2 20 = 4.3)Very hard to compress.
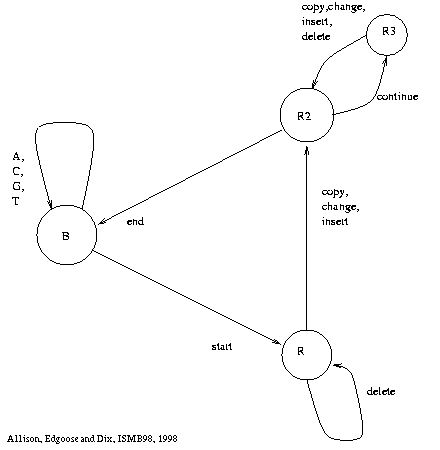
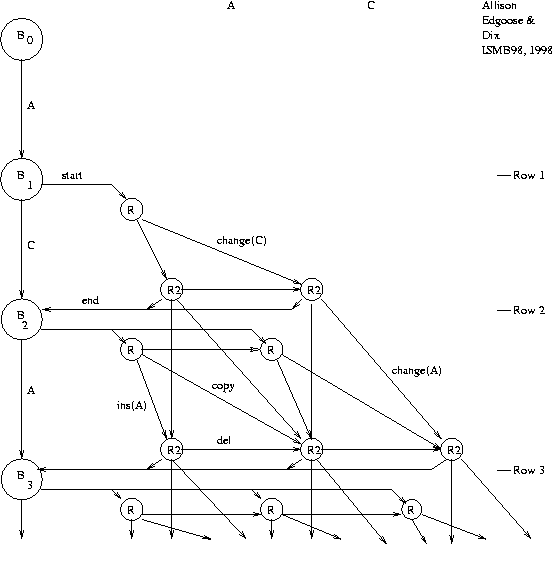
1. Approximate repeats model
(ARM )
for DNA:
<--end copy--<
base
copy
>--start copy-->
(4+, 1-)
Repeat Graph -
all explanations under ARM.
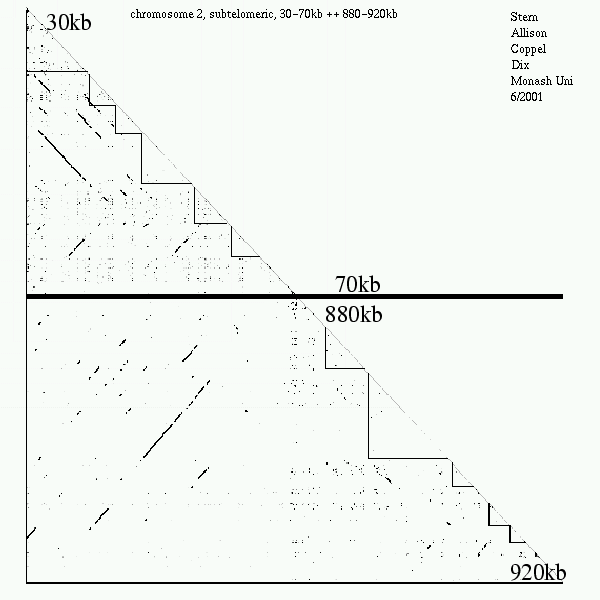
2-D Probability Plot
Note, P.f. is 80% AT.
1-D Information Content Sequence
Note,
1-D => linear space.
2. A Browser
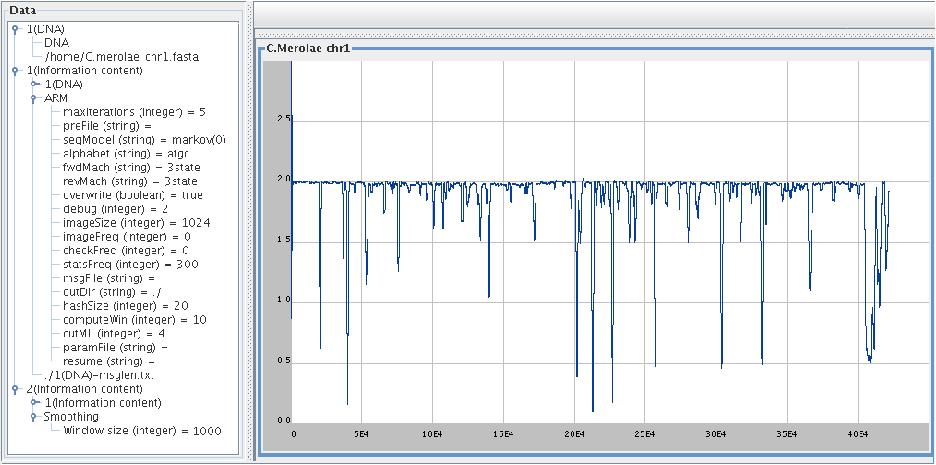
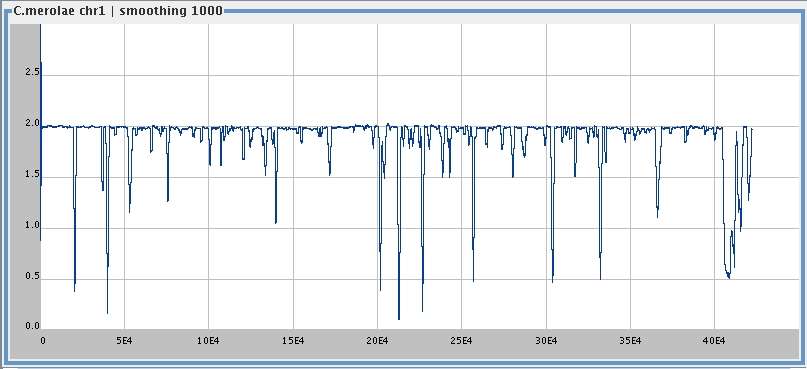
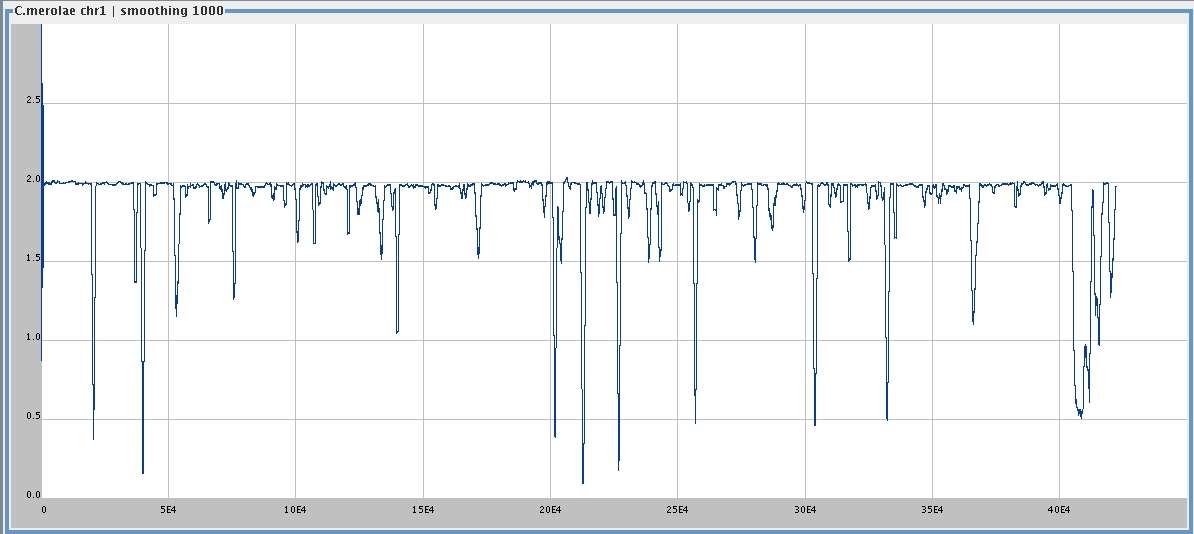
Cyanidioschyzon merolae , chromosome 1 (422,393 bp),
smoothing window 1000,
[
small ]
[
med ]
[
big ]
Differencing
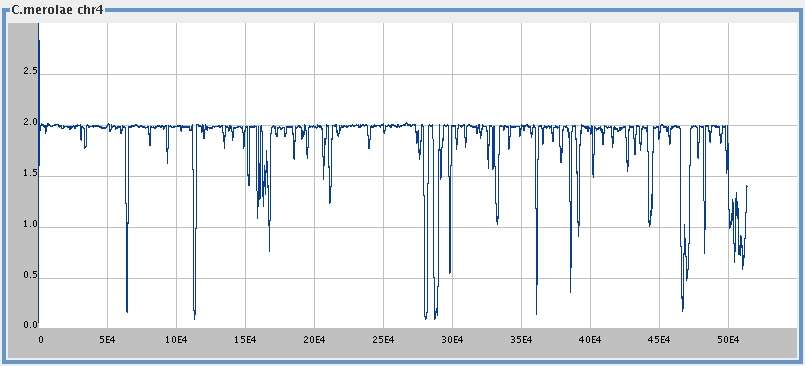
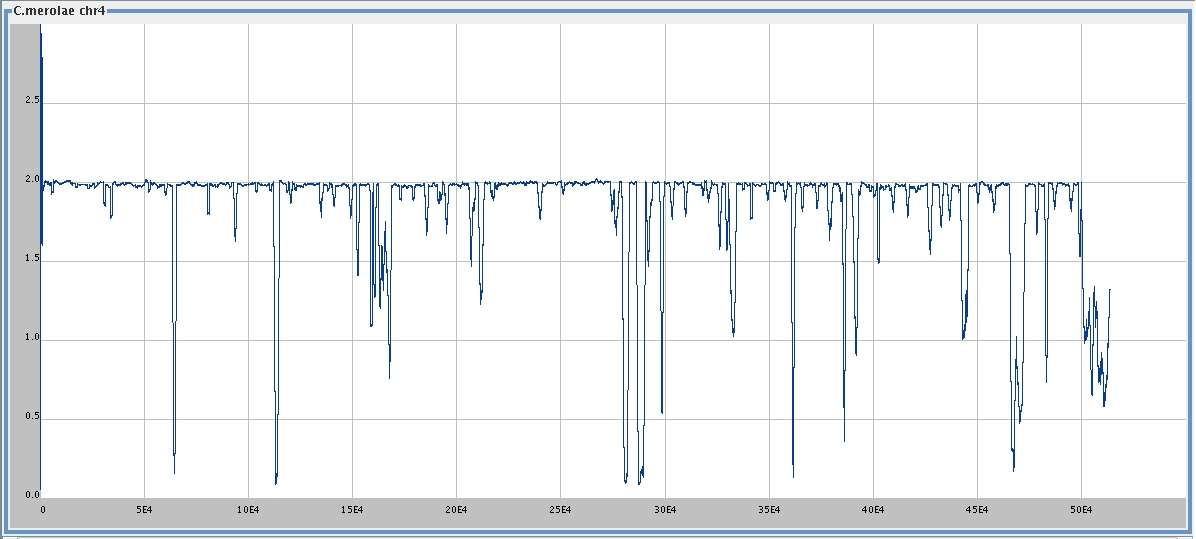
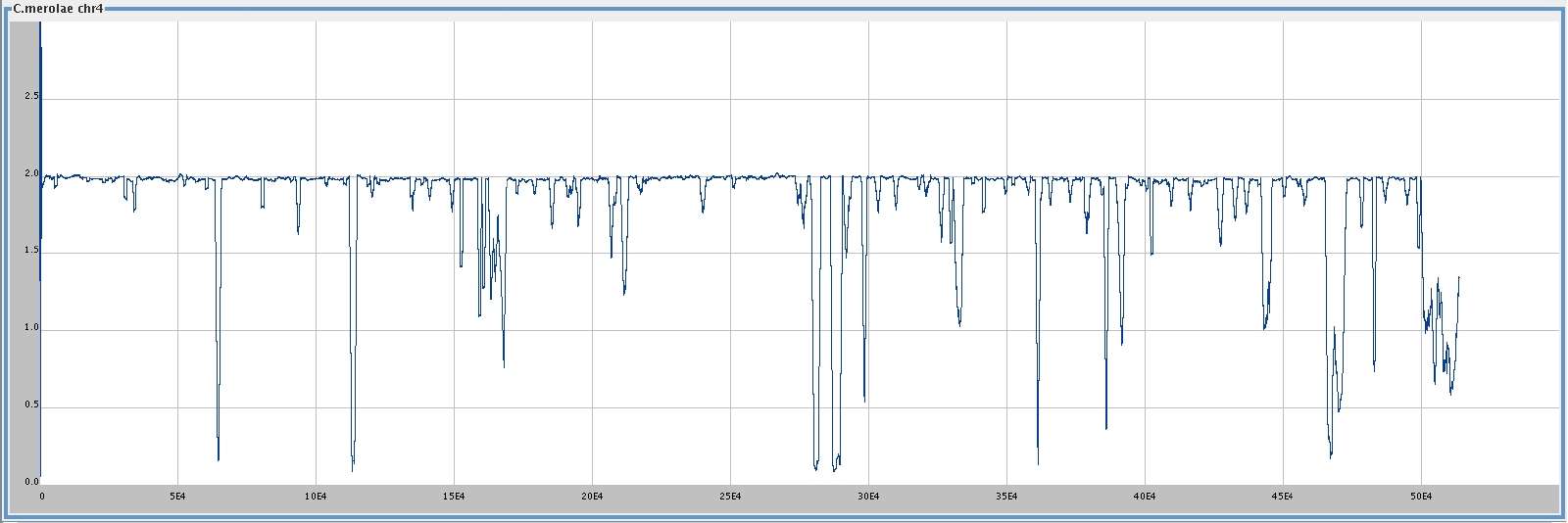
C. merolae chromosome 4 (513,790 bp),
[
med ]
[
big ],
and . . .
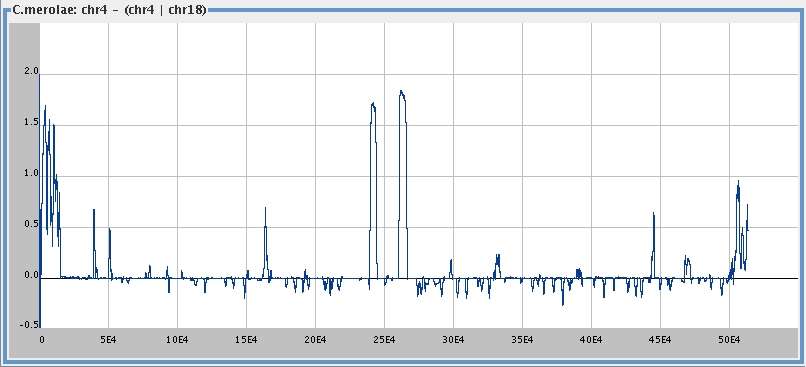
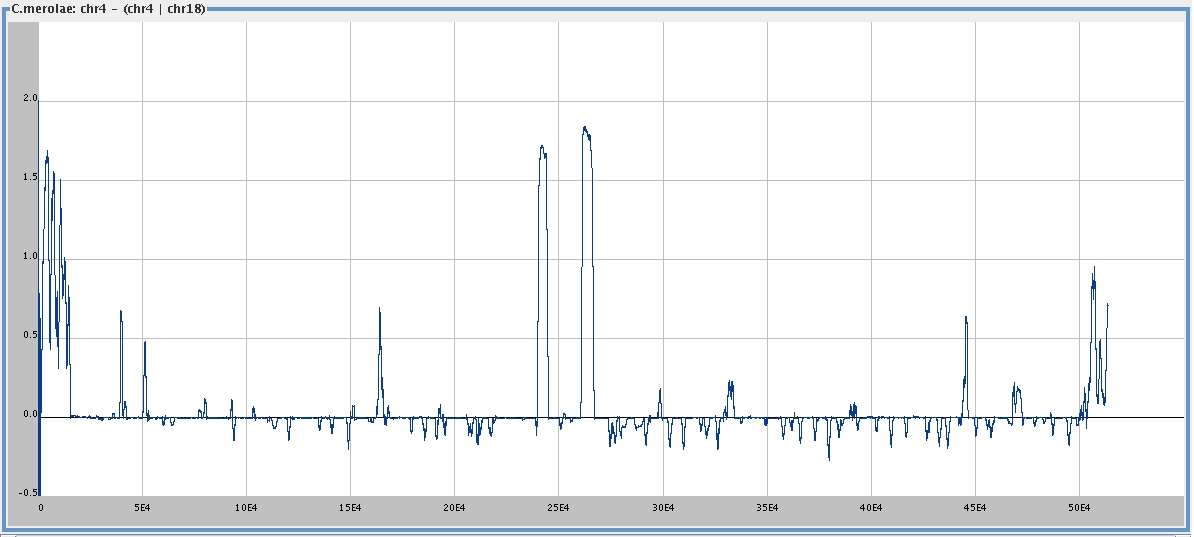
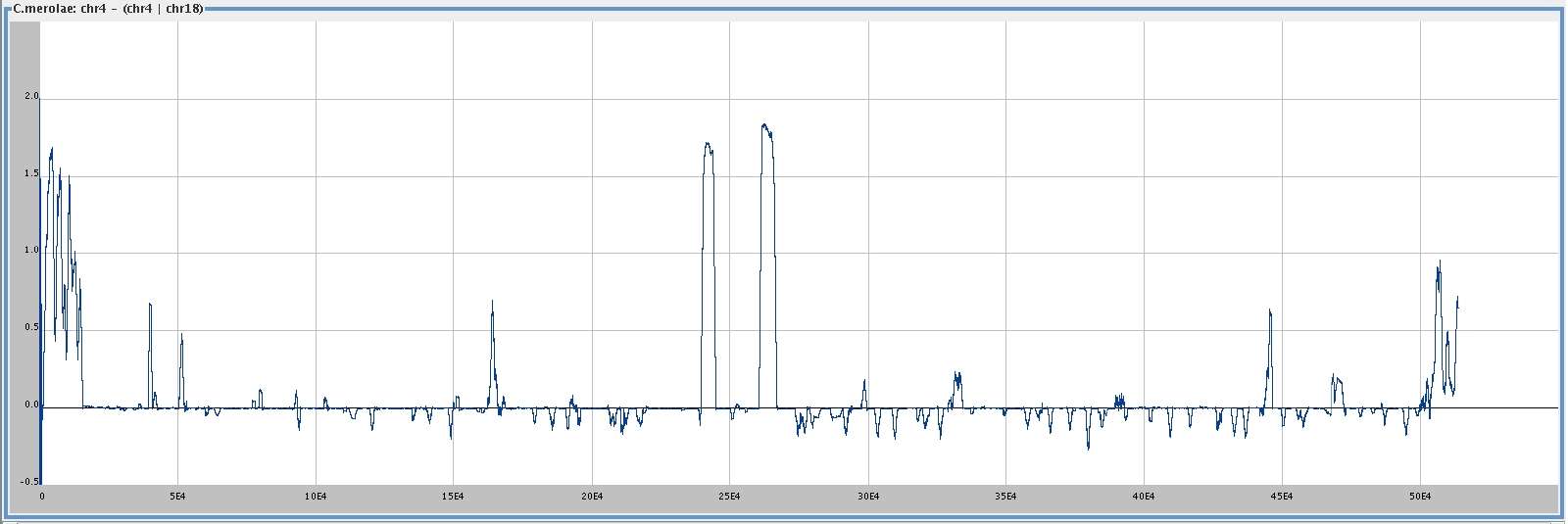
. . . its difference from
chromosome 4 given 18,
smoothing window 1000,
I (c4) - I (c4 |
[
med ]
[
big ]
Two or more sequences
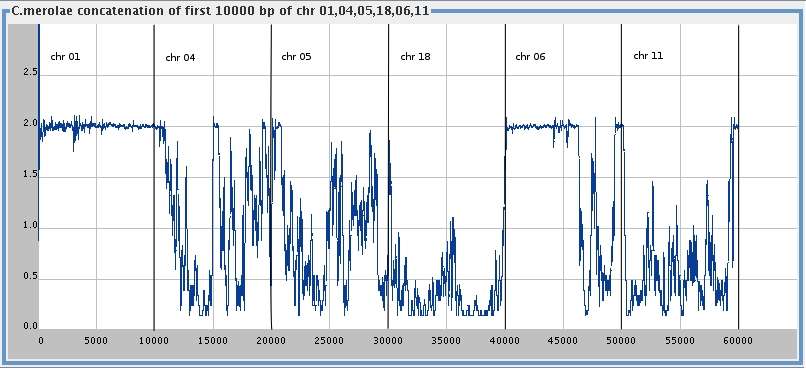
C. merolae 10,000 bp for
chromosomes
1* , 4* , 5* , 18* ,
6+ & 11+ , smoothing window 100
(* elt P, + elt PH),
[med ]
[big ]
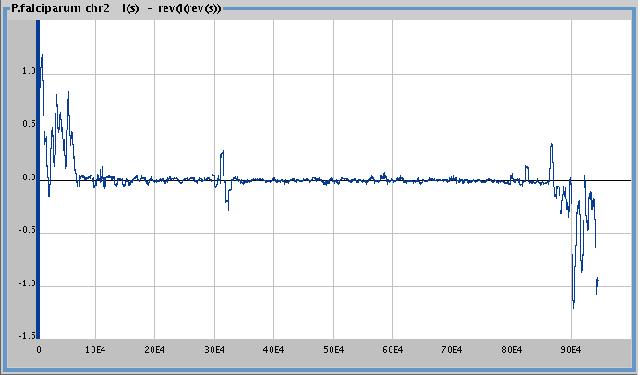
Using reversal
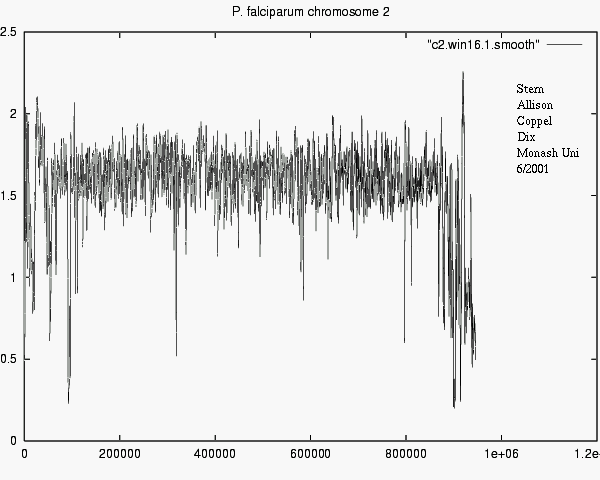
P. falciparum ,
I (chr2) - rev ( I ( rev (complement(chr2))))
3. Experts model (XM)
A blend (weighted average) of: A global background expert, e.g., a low-order MM. A local context expert, e.g., a "windowed" MM. Forward copy experts (offset, prob. copy v. change). Reverse complement copy experts (ditto). Good compression,
simple,
information per-symbol,
fast.
XM on DNA, e.g.,
BioC
GenC
DNAC
DNAP
CDNA
ARM
XM
HUMHBB
1.88
1.82
1.79
1.78
1.77
1.73
1.75
bits per baseXM on protein, e.g.,
CP
ProtComp
LZ-CTW
XM
SC,
4.15
3.94
3.95
3.89
bits per amino acid
4. Low-information substrings and alignment
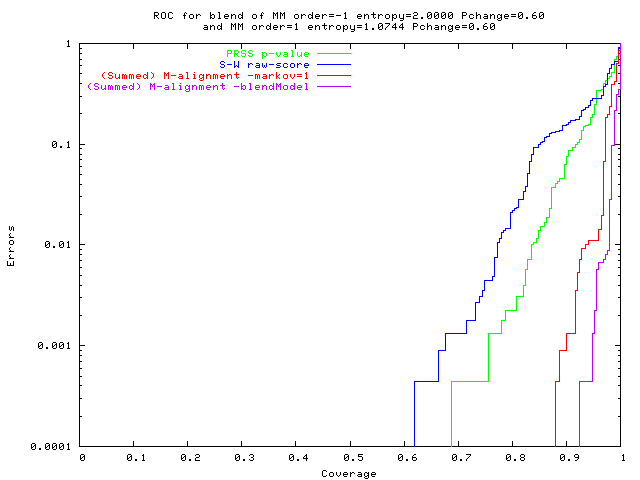
Low-information ~ repetitive ~ compressible. Cause false +ve matches for standard software, so . . . (i) sometimes "masked-out" (discarded), and/or (ii) significance test: shuffle & re-align (PRSS program).
Build a sequence-model into
the alignment algorithm. Fewer false +ves, and false -ves, than PRSS and Blast on real and
artificial test data. Time-complexity unchanged (but worse constant).
. . .
m_align ( seq_1, model_1 , seq_2, model_2 ) { ... };
Gives a natural significance test for given propulation model(s). Changes rank-order of alignments cf. "uniform" model,
as it should (rather than est. significance of a bad alignment!). model_i can be almost any model,
e.g., ARM, or XM.
. . . ROC curves
Conclusions
Calculating information carefully,
using it,
not discarding it,
gives accurate results,
few false +ves or -ves.
Information content sequences are 1-D, linear-space.
Many operations on them take linear-time.
Feasible for millions of bases.
Also see
L. Allison, T. Edgoose, T. I. Dix.
Compression of Strings with Approximate Repeats Intell. Sys. in Molec. Biol.,
pp.8-16, 1998 .Discovering patterns in Plasmodium falciparum genomic DNA Molecular and Biochemical Parasitology,
118 (2), pp.175-186, 2001 .Modelling alignment for non-random sequences Springer Verlag, LNCS Vol.3339,
pp.203-214, 2004 .Comparative Analysis of Long DNA Sequences by Per Element Information Content Using Different Contexts BMC Bioinformatics, 8 (Suppl 2):S10,
May 2007 .A Simple Statistical Algorithm for Biological Sequence Compression DCC 2007, pp.43-52, March 2007 .Bioinformatics ]
[MML ]
[interests ].
©
Lloyd Allison
Computer Science and Software Engineering,
Monash University, Australia 3800.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}